-
Overview
-
Sequence Input
-
Database Search
-
Multiple Alignment
-
Key Annotation
-
Structure Input
-
Paired
-
Tools
-
Miscellaneous
-
Statistics
-
Licence File
Key Annotation
Key Annotation is the term used in abYsis for numbering and annotating sequences. Only sequences successfully numbered can be fully annotated.
Classification and Numbering
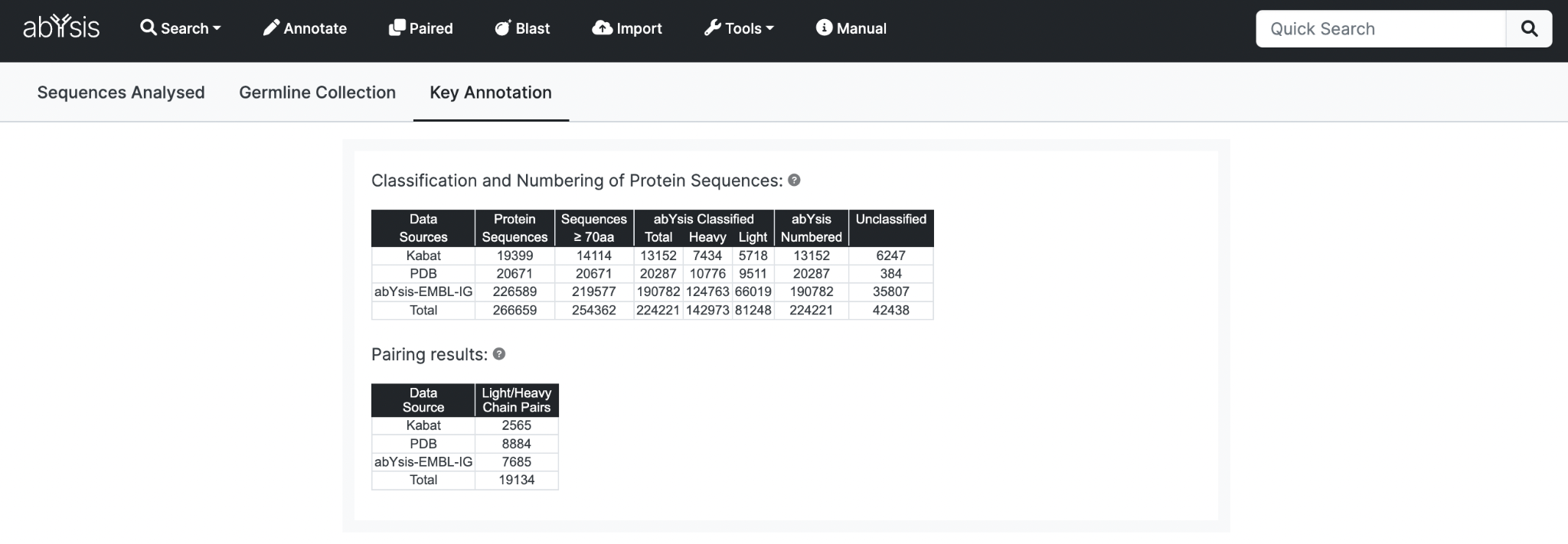
The upper table shows classification and numbering statistics for Protein Sequences in abYsis.
Data from public databases is assessed by a combination of textual information from the Data Source and abYsis analysis.
Both are used to maximise coverage. Occasionally textual information is ambiguous whilst sometimes sequence analysis fails to provide a classification. Where there is an inconsistency, the sequence analysis result is preferred, and a warning is recorded for the sequence.
The table columns are:
- Data Source Origin of sequences
- Protein Sequences Total number of protein sequences processed. Same as the total in the Sequences Analyzed tab.
- Sequences ≥70aa Total after discarding short sequences and fragments. A meaningful length of protein sequence is required for automated systems to work.
- abYsis Classified – Total sequences classified as either Light or Heavy chains.
- abYsis Classified – Heavy sequences classified.
- abYsis Classified – Light sequences classified.
- abYsis Numbered Sequences successfully Classified and Numbered. Protein sequences shorter than 70 residues are not processed through the numbering pipeline.
- Unclassified Sequences not successfully classified as heavy or light.
Pairing results
Number of successfully paired Light/Heavy chains (i.e. complete antibodies) from each data source.
abYsis attempts to pair Heavy and Light chains from public databases by combining the classification results described above with additional textual information from the data source:
- antibody 'name'
- clone identifier
- specificity
- publication details.
Where structural information is available from PDB entries, an analysis of interface residues is made, to identify the paired conformations.