-
Overview
-
Sequence Input
-
Database Search
-
Multiple Alignment
-
Key Annotation
-
Structure Input
-
Paired
-
Tools
-
Miscellaneous
-
Statistics
-
Licence File
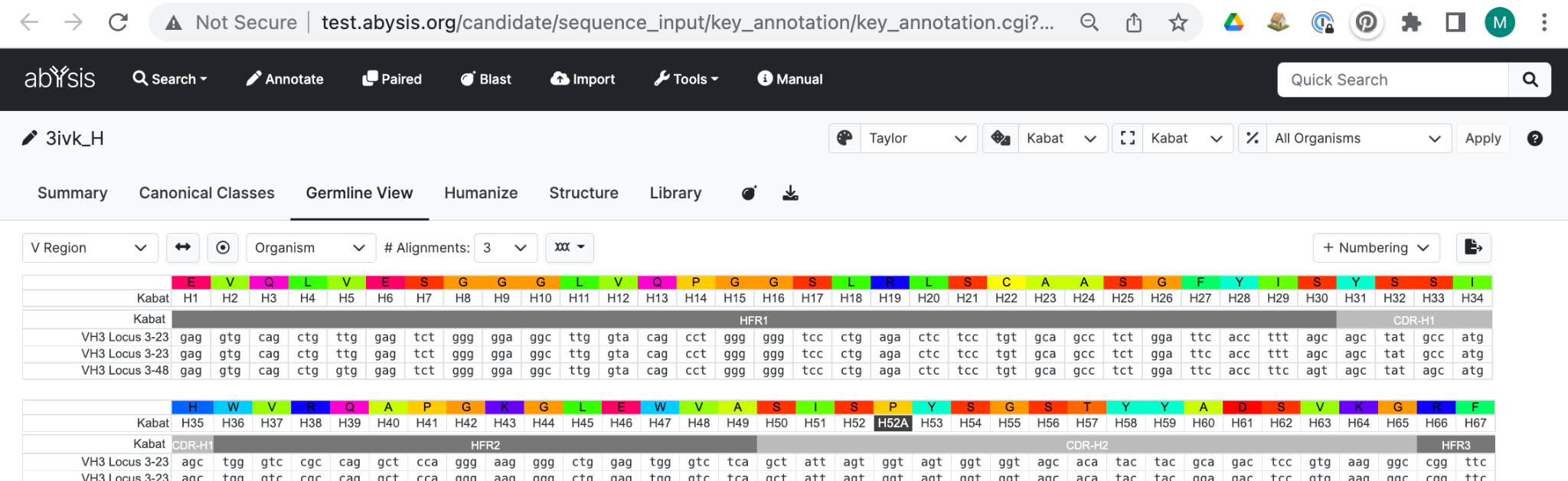
Germline
This tab shows alignments to the most similar V, D and J germline gene segments in abYsis.
Underlying searches always use the protein sequence as the query and germline DNA as the target.
Annotation specific to this Tab show the closest Germline Sequences. abYsis provides Human and Mouse germline data from NCBI and the VBASE (Medical Research Council). Commercial users can upload Germline data from other own sources that they may have access to.
For each row in the graphic there is a corresponding row in the Table below with additional information.
Menus specific to the Germline Tab are
Region - determines if only the V region or whether the alignment view focuses on particular segments (V, D or J), junction regions (V-J or V-D-J) or an overview showing the whole sequence at once.
Full Width Display - Allows the sequence to be toggled between in single fragment view and the default view.
Amino Acid Translation - translates the Germline DNA in the reading frame of the alignment.
Organism - which will restrict display to the chosen organism enabling, for example, the top human germline sequence hits to a sequence.
#Alignments which controls the number of alignments displayed
Germline Tools which provides access to advanced options
In the table, Identities shows the total number of nucleotide and/or amino acid matches between the query and the aligned germline DNA sequence. Where query codons are known, we count 1 for each nucleotide match. Where query codons are not known we count 1 for each amino acid match. The second number in the Identities column is the maximum possible number of identities over the aligned region, counted in this way. In default mode, the targets are ranked in order of decreasing identity.
Germline Tools
You can set the gap penalty in the Advanced Options when the Gaps checkbox is checked. When unchecked it will use a gap penalty of -1400
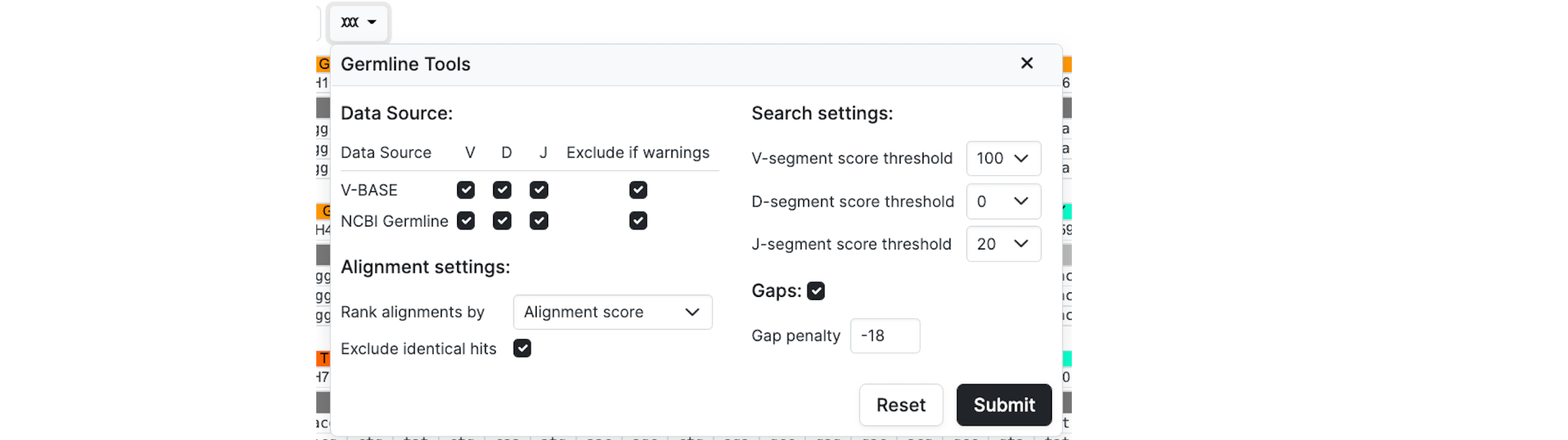
Data Source allows you to control the target germline sequences to search. You may specify the Data Source, Gene Segment and exclude target sequences that have Warnings.
Alignment settings allows you to Rank alignments with targets by Alignment Score (the default) or by Identity. You can also Specify whether or not to Exclude identical hits - i.e. germline sequences with identical sequences - in the results set.
Search settings set scoring thresholds for alignment with different gene segments to be specified.
Gaps allows the gap penalty to be set. The default is -18. Values above -10 are not allowed because of the exhaustive system requirements required to run the analysis.
Methods
abYsis uses the exonerate suite of programs [Slater and Birney (2005) BMC Bioinformatics 6:31] for underlying Protein/DNA sequence comparisons.
The V segments are searched first, then the J segments. If V and J hits are obtained and the chain is a heavy chain, the short region between the top hitting V and J aligned regions is excised and used to make a final exhaustive search of the short D segment sequences. Search thresholds may be adjusted using the Search Settings dropdowns.