-
Overview
-
Sequence Input
-
Database Search
-
Multiple Alignment
-
Key Annotation
-
Structure Input
-
Paired
-
Tools
-
Miscellaneous
-
Statistics
-
Licence File
Blast
The NCBI Blast family of programs are implemented within abYsis. The flavour of Blast run depends on your query and database:
Blast Programs
blastp
compares an amino acid query sequence against a protein sequence database
blastn
compares a nucleotide query sequence against a nucleotide sequence database
blastx
compares a nucleotide query sequence translated in all reading frames against a protein sequence database
tblastn
compares a protein query sequence against a nucleotide sequence database dynamically translated in all reading frames
tblastx
compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database. Please note that tblastx program cannot be used with the nr database on the Blast Web page.
Blast Parameters
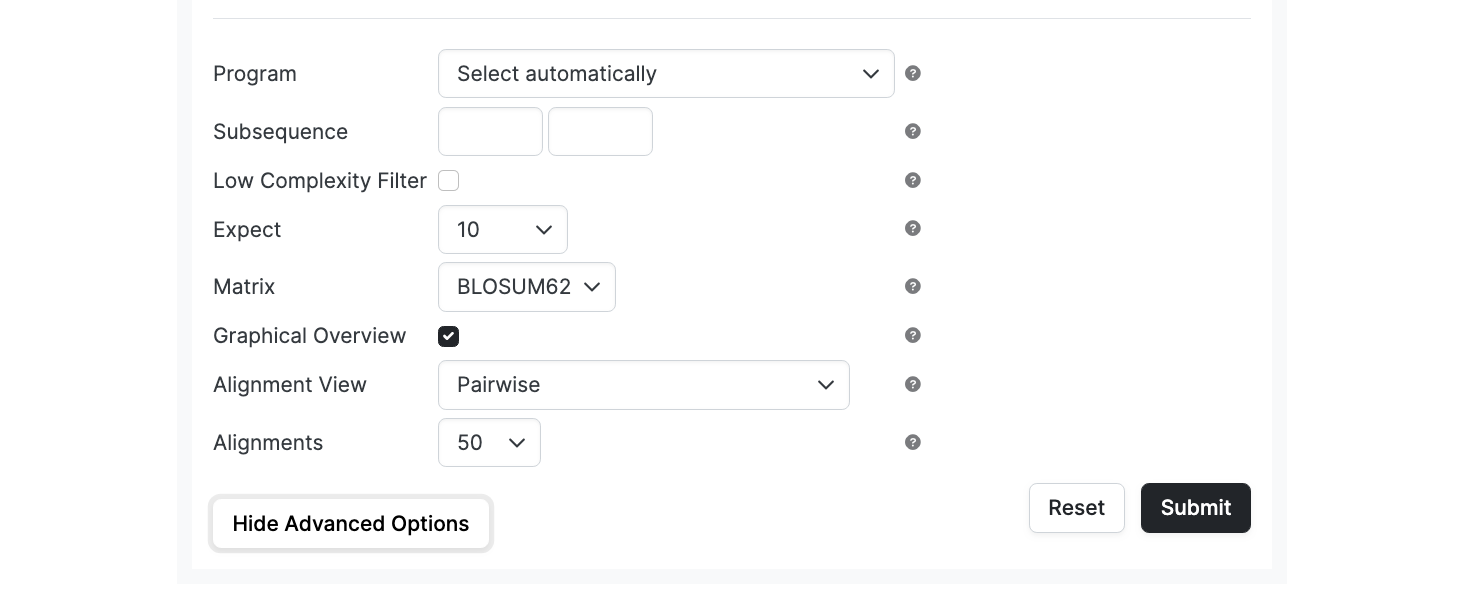
Subsequence
Only uses the selected range of the query sequence. Enter from and to coordinates to define the subsequence.
Low Complexity Filter
Mask off segments of the query sequence that have low compositional complexity, as determined by the SEG program of Wootton & Federhen (1993) [Computers and Chemistry 17:149-163] or, for BLASTN, by the DUST program of Tatusov and Lipman (Unpublished, NCBI/Toolkit).
Filtering can eliminate erroneous hits in large databases. Filtering is only applied to the query sequence (or its translation products), not to the database sequences as they have already been qualified to be probable antibody sequences in order to be loaded in to abYsis.
Expect
The statistical significance (e-value) threshold for reporting matches against database sequences; the default value is 10, such that 10 matches are expected to be found merely by chance, according to the stochastic model of Karlin and Altschul (1990) [Proc Natl Acad Sci U S A. 87:2264-2268]. If the statistical significance ascribed to a match is greater than the Expect threshold, the match will not be reported. Lower Expect thresholds are more stringent, leading to fewer chance matches being reported.
Matrix
The Matrix options are described in the separate Matrix Help Section.
Alignment View
A selection of alignment output formats are available. Click the Alignment View dropdown to select an output style. The default viewer is JSAV, the JavaScript Sequence Alignment Viewer that is also used to view Search results and multiple-sequence Key Annotations. 
See Blast Search for more information.
Descriptions
Restricts the number of short descriptions of matching sequences reported to the number specified [default: 100]. See also Expect.
Alignments
Restricts database sequences to the number specified for which high-scoring segment pairs (HSPs) are reported default: 50]. If more database sequences than this happen to satisfy the statistical significance threshold for reporting (see Expect, above), only the matches ascribed the greatest statistical significance are reported.